Blog Details
Blog Details
BOIN vs 3+3: Why Model-Based Dose Escalation is Winning FDA Approval in the Project Optimus Era
BOIN vs 3+3: Why Model-Based Dose Escalation is Winning FDA Approval in the Project Optimus Era
This Article — 8-Minute Read
This Article — 8-Minute Read


The End of "Higher is Better" in Oncology Drug Development
Nearly 50% of targeted cancer therapies approved in the past decade have required post-market dose reductions. The culprit? An outdated approach to finding the right dose.
For decades, the 3+3 design ruled Phase I oncology trials. It was simple, conservative, and built for an era when cytotoxic chemotherapy dominated cancer treatment. But modern oncology has moved on. Targeted therapies, antibody-drug conjugates, and immunotherapies behave differently. They often achieve maximum efficacy at doses well below what patients can tolerate.
The FDA recognized this mismatch and launched Project Optimus, fundamentally changing what regulators expect from early-phase oncology programs. At the center of this shift is a statistical methodology that biotechs can no longer afford to ignore: the Bayesian Optimal Interval (BOIN) design.
What Project Optimus Means for Your Phase I Trial
In August 2024, the FDA published final guidance that explicitly discourages reliance on finding the Maximum Tolerated Dose through rule-based escalation like 3+3. Instead, sponsors must now demonstrate comprehensive dose optimization aimed at identifying an Optimal Biological Dose.
The implications are significant:
Expanded safety assessment. The FDA now requires sponsors to capture low-grade symptomatic toxicities and patient-reported outcomes that impact long-term tolerability, not just acute severe events.
Dose-response characterization. Sponsors must integrate pharmacokinetic and pharmacodynamic data early to justify their selected dose before pivotal trials begin.
Randomized dose evaluation. The guidance strongly recommends parallel dose comparisons during development to reduce bias in dose-exposure relationships.
Most critically, the FDA has warned it may place trials on clinical hold if sponsors propose doses inadequately supported by robust optimization data. The stakes have never been higher.
Why the 3+3 Design is Failing Modern Oncology
The 3+3 algorithm became standard because clinicians could execute it at the bedside without statistical software. Enroll three patients. If no one experiences a dose-limiting toxicity, escalate. If one does, expand to six. If two or more experience toxicity, stop and declare the previous dose as the MTD.
Simple, but fundamentally flawed.
Short memory. Decisions rely only on the current cohort of three to six patients, ignoring valuable data from previous dose levels.
Arbitrary sample size. Capping evaluation at six patients per dose creates high statistical uncertainty. Random variation frequently drives incorrect MTD identification.
Chronic underestimation. Conservative stopping rules routinely identify an MTD below the true optimal dose, relegating trial participants to sub-therapeutic levels.
Poor allocation efficiency. A large proportion of patients receive the lowest doses, limiting the safety and efficacy data available at potentially therapeutic levels.
For cytotoxic drugs where toxicity and efficacy both increase with dose, these limitations were tolerable. For modern targeted agents where the relationship is more complex, they are disqualifying.
BOIN: Statistical Rigor Without the Complexity
The Bayesian Optimal Interval design solves the central challenge facing oncology biostatistics: how to bring rigorous statistical methods to clinical trials without requiring real-time software intervention or opaque algorithms.
BOIN is classified as a "model-assisted" design. It uses Bayesian decision theory to calculate optimal escalation and de-escalation boundaries before the trial begins. These boundaries are printed in the protocol as a simple lookup table. Site staff make dose decisions exactly as they would with 3+3, but with dramatically better statistical properties.
How it works:
The design establishes a target DLT rate based on the drug's expected toxicity profile, typically 20%, 25%, or 30%. Using this target, BOIN calculates two boundaries:
Escalation boundary: If the observed DLT rate falls below this threshold, escalate to the next dose.
De-escalation boundary: If the observed DLT rate exceeds this threshold, reduce to the previous dose.
Between boundaries: Retain the current dose for the next cohort.
BOIN also incorporates formal overdose control. If statistical evidence suggests even the lowest dose exceeds acceptable toxicity with high probability, the trial stops immediately, protecting patients from continued exposure to an unsafe agent.
Head-to-Head: BOIN Outperforms 3+3 on Every Metric
Extensive simulation studies leave no doubt about which design performs better.
Metric | 3+3 Design | BOIN Design |
|---|---|---|
MTD Identification Accuracy | Routinely underestimates true MTD | 12-16% higher correct selection rate |
Patient Allocation | Many patients at sub-therapeutic doses | Clusters enrollment around true optimal dose |
Overdose Control | Stops quickly but at expense of trial success | Formal Bayesian boundaries balance safety and efficacy |
Sample Size Flexibility | Rigid cap of 6 patients per dose | Dynamic cohort sizes up to total sample limit |
Trial Speed | Mandatory waiting between cohorts | TITE-BOIN extension allows continuous enrollment |
In scenarios where the true MTD sits at the highest proposed dose level, BOIN is nearly three times more likely to correctly identify that dose compared to the conservative 3+3 approach.
The Business Case: Faster Timelines, Lower Risk
Phase I oncology trials routinely cost $4 million to $15 million. The financial case for BOIN extends well beyond upfront design costs.
Preventing downstream failure. By accurately identifying the optimal dose, BOIN dramatically reduces the risk of advancing a sub-therapeutic or excessively toxic dose into Phase II and III. A failed pivotal trial from poor dose selection can cost hundreds of millions.
Accelerating enrollment. Traditional 3+3 creates accrual bottlenecks. Sites must wait 21 to 28 days for the DLT observation window to close before enrolling the next cohort. The Time-to-Event BOIN extension (TITE-BOIN) incorporates survival analysis techniques that allow continuous enrollment even while previous patient data remains pending. This can shorten Phase I timelines by months.
Satisfying PK/PD requirements. Project Optimus demands robust pharmacokinetic and pharmacodynamic data across multiple dose levels. The Backfilling BOIN extension (BF-BOIN) allows sponsors to assign patients to cleared lower doses specifically to generate this data without stalling the primary escalation.
What FDA Reviewers Expect in Your IND
The BOIN methodology received official "Fit-for-Purpose" designation from the FDA's Office of Biostatistics following rigorous review. This pre-validation significantly de-risks adoption for sponsors.
However, utilizing an endorsed framework does not exempt sponsors from thorough documentation. FDA statistical reviewers expect:
Justified target DLT rate. The entire BOIN boundary structure depends on this variable. Defaulting to 30% because that was standard for cytotoxic trials is a common mistake. For targeted agents or earlier-stage disease settings, 20% or 25% may be more appropriate.
Comprehensive simulation report. Reviewers require evidence that the design performs well across scenarios, including stress tests where all doses are highly toxic or the dose-toxicity curve is flat.
Explicit prior distributions. For any Bayesian elements, sponsors must justify their mathematical assumptions and demonstrate that reasonable alternative assumptions do not substantially change conclusions.
Structured backfilling protocols. Ad hoc patient assignment to lower doses can invalidate the trial's statistical architecture. Formal extensions like BF-BOIN must be prospectively specified.
The Regulatory Landscape: FDA and EMA Alignment
The European Medicines Agency has instituted parallel expectations in its CHMP Revision 7 guidelines on anticancer medicinal products. Like the FDA, the EMA recognizes that dose optimization must extend beyond acute tolerability.
Key EMA requirements include:
Early pharmacokinetic and pharmacodynamic assessment to drive dose selection
Mechanistic characterization of both on-target and off-target toxicities for combination therapies
Integration of patient-reported outcomes and quality of life measures into early-phase development
The EMA accepts Bayesian adaptive designs when sponsors provide transparent operating characteristics demonstrating reduced bias and improved interpretability. Biotechs pursuing both FDA and EMA approval can align their development strategy around BOIN without regulatory friction.
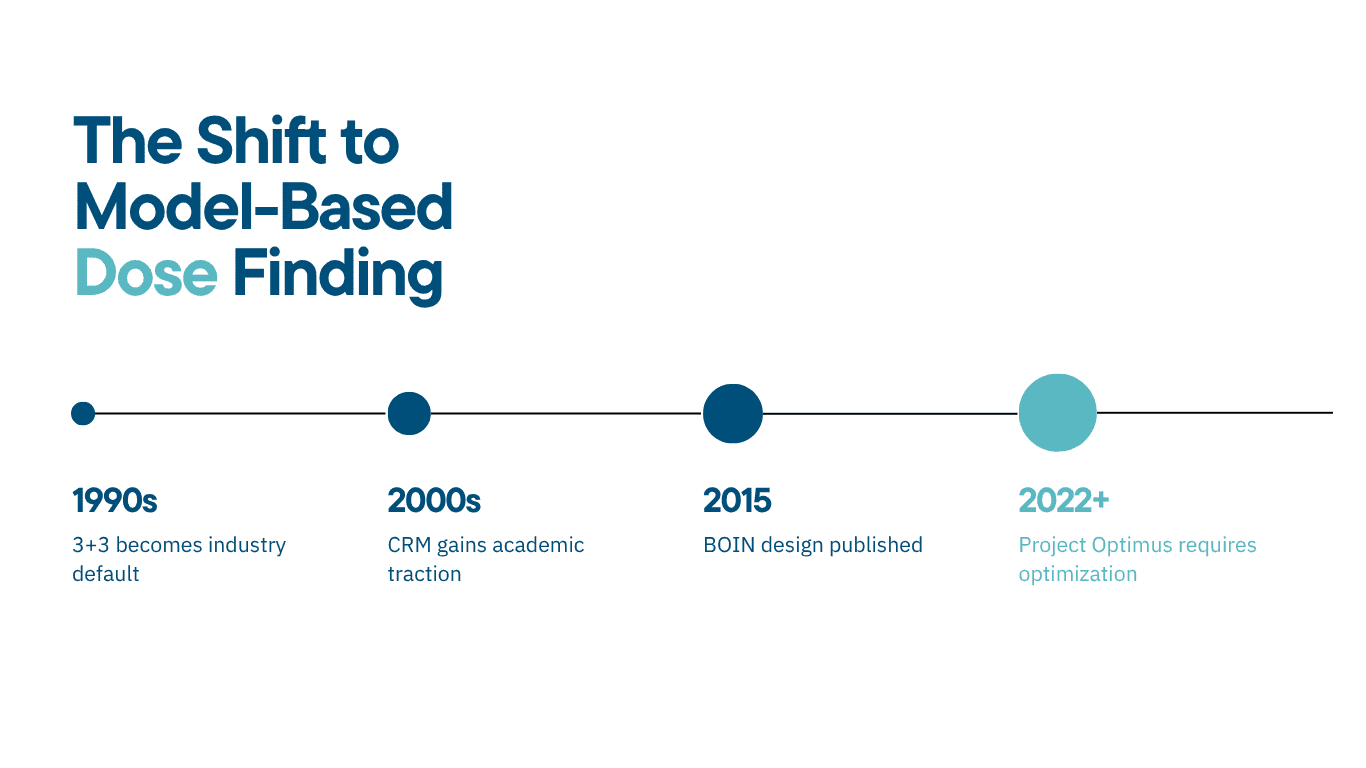
Industry Adoption: Who is Using BOIN?
The shift is measurable. A longitudinal analysis of 367 industry-sponsored Phase I oncology protocols between 2021 and 2024 found that Bayesian design adoption increased from 48% to 75%. The 3+3 approach is in rapid decline among sophisticated sponsors.
Notable implementations include:
Corbus Pharmaceuticals utilized BOIN for CRB-701, a Nectin-4 targeting ADC, presenting safety and PK data generated through the design at ESMO.
FB849 developers deployed adaptive BOIN in Phase Ia monotherapy before transitioning to BOIN-guided combination cohorts with pembrolizumab.
DB-1317 sponsors combined accelerated titration for initial low-dose cohorts with formal BOIN for subsequent escalation once safety signals emerged.
These companies are publicizing their adoption of advanced statistical methods as signals of regulatory sophistication to investors, clinical partners, and potential acquirers.
Making the Transition
Clinging to the 3+3 design in the current regulatory environment represents a profound strategic risk. Dr. Richard Pazdur, Director of the FDA Oncology Center of Excellence, characterized it directly: developing a drug program without thorough dose understanding is "akin to building a house on quicksand."
The transition to BOIN does not require abandoning operational simplicity. Clinical site staff use the same decision-making process they always have. They simply reference a pre-calculated boundary table instead of a 3+3 flowchart.
What changes is the statistical foundation underlying those decisions, and the probability that your trial will identify the right dose for your molecule.
Partner With Specialists Who Understand Modern Dose-Finding
Navigating Project Optimus, FDA IND submissions, and Bayesian trial design requires deeply specialized biostatistical expertise. The difference between a well-designed BOIN protocol and an inadequate one may determine whether your program advances to pivotal trials or stalls at Phase I.
OncoMetrika provides oncology-focused biostatistics consulting for biotechs and pharmaceutical teams designing Phase I trials. We specialize in model-based dose escalation, simulation-based protocol development, and regulatory strategy for FDA and EMA submissions.
Schedule a Strategy Call to discuss your Phase I design challenges. We will review your asset, your regulatory pathway, and how modern dose-finding methods can position your program for success.
Our Recent Blog



Your cancer trial data deserves more than a generalist.
Whether you need a statistical analysis plan for your Phase II solid tumor study or a network meta analysis for your NICE submission, let's talk.
Your cancer trial data deserves more than a generalist.
Whether you need a statistical analysis plan for your Phase II solid tumor study or a network meta analysis for your NICE submission, let's talk.
Your cancer trial data deserves more than a generalist.
Whether you need a statistical analysis plan for your Phase II solid tumor study or a network meta analysis for your NICE submission, let's talk.